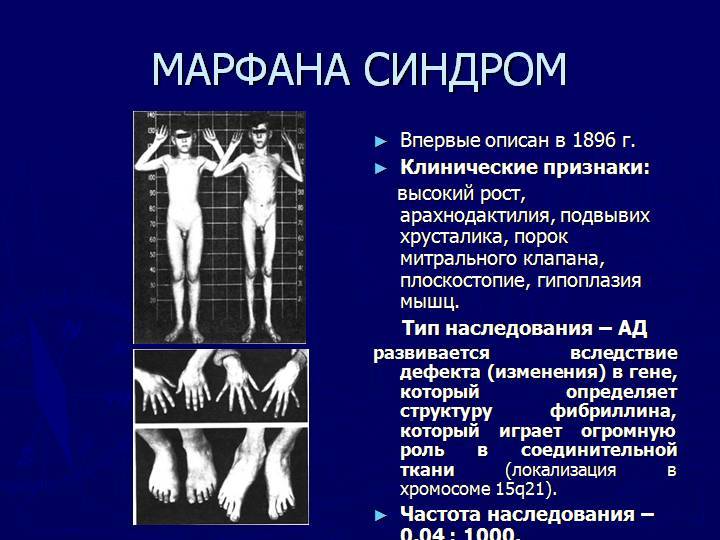
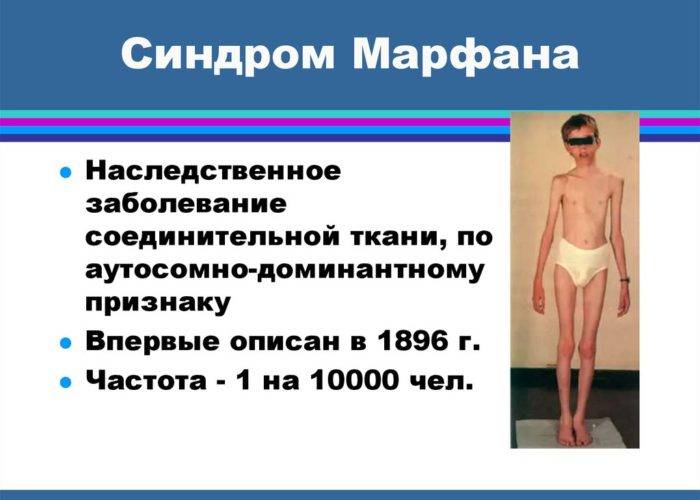
Симптомы Болезни (синдрома) Марфана:
Клинические симптомы заболевания разделяют на несколько групп, которые отражают точную локализацию проявлений соединительнотканной (мезенхимальной) дисплазии:
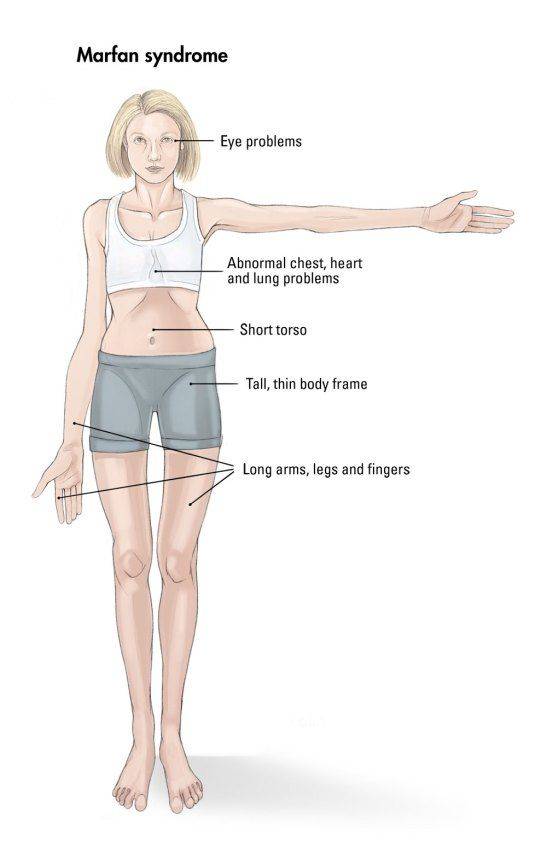
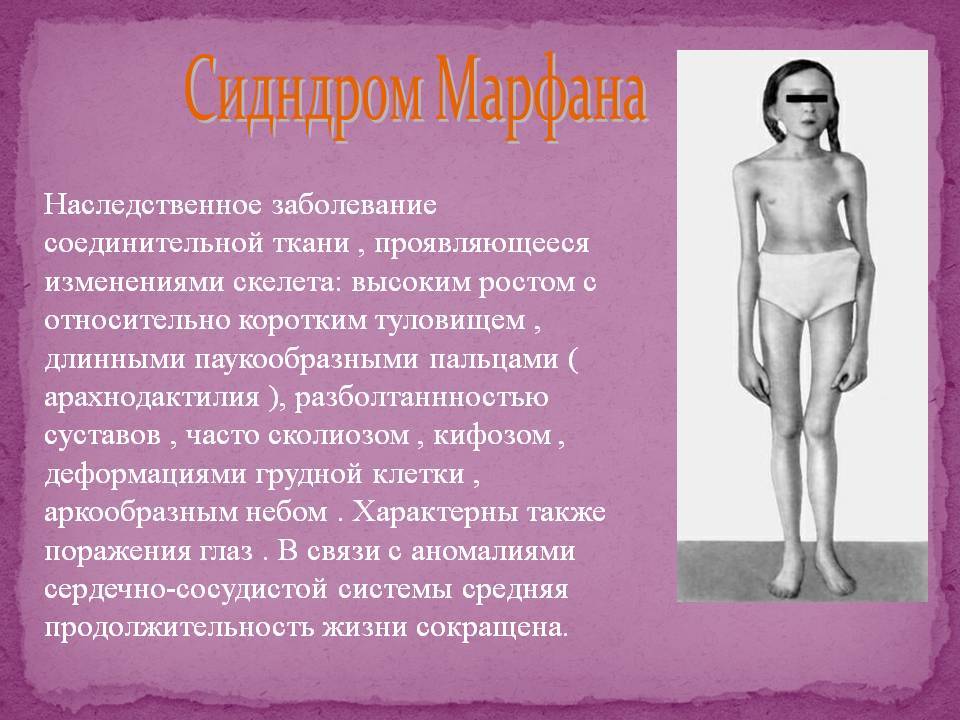
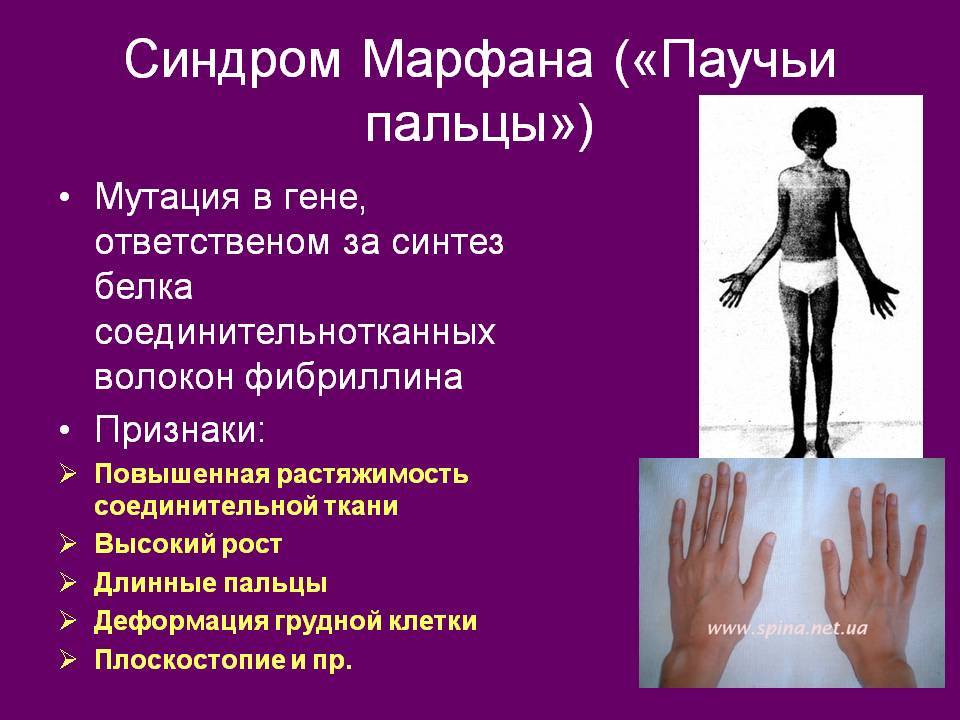
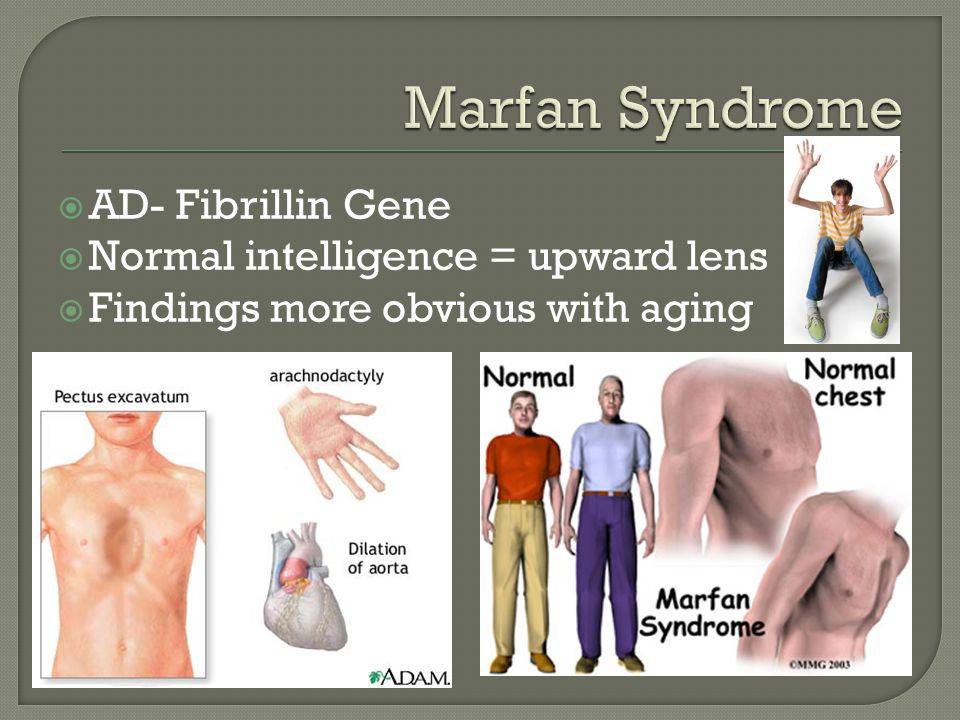
- костно-суставные расстройства (астеническая конституция, узкий лицевой череп с «птичьим» выражением лица, плоскостопие, узкая и деформированная, килевидная или воронкообразная грудная клетка, арахнодактилия кистей и стоп, кифосколиотическая деформация позвоночника, гипермобильность суставов и сухожилий). Костно-суставные нарушения есть у большинства больных;
- изменения мягких тканей (малая масса тела, мышечная гипотония, гипоплазия жировой ткани и мускулатуры, плоскостопие);
- изменения внутренних органов (аневризма восходящей аорты, клапанные пролапсы, особенно пролапс митрального клапана, гипоплазированное, расширение корня аорты и легочной артерии, аневризмы синусов Вальсальвы, «висячее», «капельное» сердце, уменьшение долей легких, слишком длинный и гипопластичный кишечник);
- нарушения в системе зрения (голубые склеры, гиперметропия высоких степеней, аниридия, выраженная миопия, эктопия и подвывихи хрусталика, афакия, колобома);
- расстройства центральной нервной системы (анизокория, нистагм, асимметрия сухожильных рефлексов, пирамидные расстройства);
- Расстройства гипофизарно-адреналовой системы (высокий рост, акромегалоидные расстройства, несахарный диабет, арахнодактилия, удлиненные конечности, увеличенные ступни, вегетативные расстройства);
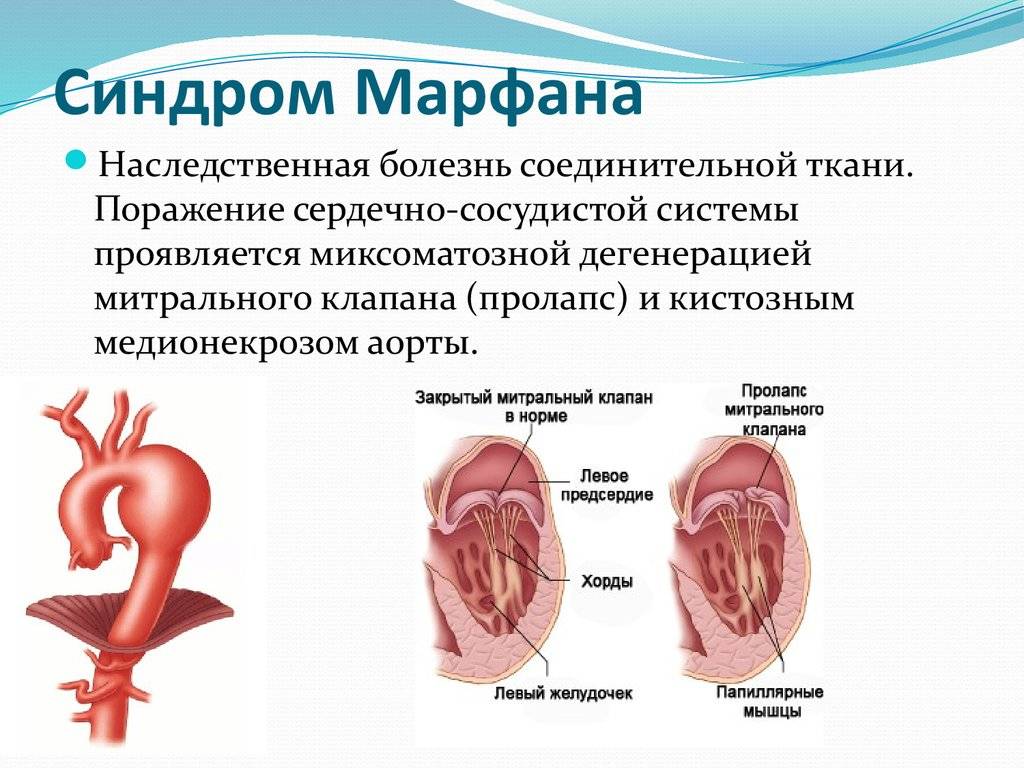
- нарушения сердечно-сосудистой системы (нарушения внутрижелудочковой проводимости, умеренные признаки гипертрофии миокарда левого желудочка и предсердия, изменения в сердце и магистральных сосудах, аортальная недостаточность, пролапс митрального клапана, нарушения внутрисердечной гемодинамики, расширение корня аорты, двустворчатый клапан аорты, митральная недостаточность, которая связана с развитием миксоматозной дегенерацией створок, увеличением их площади, расширением фиброзного кольца, удлинением хорд, «разболтанностью» створок и увеличением пролабирования). У больных наблюдается мышечная слабость и снижение активности при физических нагрузках.
Симптомы Болезни (синдрома) Марфана:
Клинические симптомы заболевания разделяют на несколько групп, которые отражают точную локализацию проявлений соединительнотканной (мезенхимальной) дисплазии:
- костно-суставные расстройства (астеническая конституция, узкий лицевой череп с «птичьим» выражением лица, плоскостопие, узкая и деформированная, килевидная или воронкообразная грудная клетка, арахнодактилия кистей и стоп, кифосколиотическая деформация позвоночника, гипермобильность суставов и сухожилий). Костно-суставные нарушения есть у большинства больных;
- изменения мягких тканей (малая масса тела, мышечная гипотония, гипоплазия жировой ткани и мускулатуры, плоскостопие);
- изменения внутренних органов (аневризма восходящей аорты, клапанные пролапсы, особенно пролапс митрального клапана, гипоплазированное, расширение корня аорты и легочной артерии, аневризмы синусов Вальсальвы, «висячее», «капельное» сердце, уменьшение долей легких, слишком длинный и гипопластичный кишечник);
- нарушения в системе зрения (голубые склеры, гиперметропия высоких степеней, аниридия, выраженная миопия, эктопия и подвывихи хрусталика, афакия, колобома);
- расстройства центральной нервной системы (анизокория, нистагм, асимметрия сухожильных рефлексов, пирамидные расстройства);
- Расстройства гипофизарно-адреналовой системы (высокий рост, акромегалоидные расстройства, несахарный диабет, арахнодактилия, удлиненные конечности, увеличенные ступни, вегетативные расстройства);
- нарушения сердечно-сосудистой системы (нарушения внутрижелудочковой проводимости, умеренные признаки гипертрофии миокарда левого желудочка и предсердия, изменения в сердце и магистральных сосудах, аортальная недостаточность, пролапс митрального клапана, нарушения внутрисердечной гемодинамики, расширение корня аорты, двустворчатый клапан аорты, митральная недостаточность, которая связана с развитием миксоматозной дегенерацией створок, увеличением их площади, расширением фиброзного кольца, удлинением хорд, «разболтанностью» створок и увеличением пролабирования). У больных наблюдается мышечная слабость и снижение активности при физических нагрузках.
Близорукость
Данное нарушение зрения проявляется более чем у половины больных. Причинами ее могут быть: шаровидная форма хрусталика, изменение преломляющей силы роговицы глаза вследствие ее уплощения и увеличения, а также быстрый рост по переднезадней оси самого глазного яблока. При этом миопия может достигать довольно высоких степеней.
У больных детей также нередко может развиваться синдром «ленивого глаза» — амблиопия. Родителям маленьких пациентов с синдромом Марфана следует быть очень внимательными к состоянию зрения ребенка. Амблиопия зачастую переходит в косоглазие или наоборот.
Диагностика
Диагноз синдром Марфана ставится на основании имеющейся клинической картины, семейного анамнеза и генетических исследований.
Процессу диагностики помогает ряд визуальных исследований. Рентгенография дает информацию о состоянии грудной клетки, изменении длины и ширины костей ладоней и пальцев. Рентгенограммы таза отражают состояние тазобедренных суставов. КТ и МРТ имеют преимущества в оценке состояния мягких тканей и детализации дефектов.
Сердечно-сосудистая система оценивается на основании физического осмотра, касающегося дефекта клапана, признаков сердечной недостаточности, ЭКГ и обычной, а в некоторых случаях чреспищеводной эхокардиографии. Состояние аорты и крупных сосудов оценивают контрастным исследованием.
Исследование зрительной системы начинается с определения остроты зрения, офтальмоскопии, осмотра глазного дна. Широко распространенным исследованием в последнее время является УЗИ глазного яблока.
Диагностика синдрома Марфана
Диагностика генетической аномалии включает в себя комплекс мероприятий по определению всех симптомов болезни, а также изучению вероятности развития мутации:
- Сбор жалоб — детальное изучение всех патологических признаков.
- Определение анамнеза — выяснение состояния здоровья родителей.
- Тщательный осмотр, измерение роста, размаха рук и других антропометрических показателей. Скрининговый тест для детей в возрасте 7–18 лет — это измерение длины среднего пальца руки. У пациентов с синдромом Марфана показатель превышает отметку в 10 см.
Генетическое обследование включает в себя выявление генотипа ДНК — идентификацию мутаций в гене FBN1. При возможности назначают специфические лабораторные тесты — определение выведения с мочой метаболитов соединительной ткани, таких как оксипролин и гликозаминогликаны.
Чтобы подтвердить нарушения развития соединительной ткани и оценить степень выраженности мутации гена FBN1, пациентам с подозрением на синдром Марфана назначают:
- ЭКГ;
- УЗИ сердца;
- КТ-ангиографию аорты и других сосудов;
- КТ грудной и брюшной полостей;
- МРТ позвоночника и головного мозга;
- специфические обследования на осмотре у офтальмолога;
- биопсию кожи.
Для окончательного определения диагноза используют общепринятые Гентские критерии 2010 года, согласно которым диагноз устанавливают в случаях:
- подтвержденной мутации гена FBN1 и расширения корня аорты или эктопией хрусталика;
- подтвержденного расширения корня аорты в сочетании с эктопией хрусталика;
- подтвержденной эктопии хрусталика в сочетании с любыми признаками системного поражения соединительной ткани.
Важно! Существует группа «марфаноподобных» синдромов, при которых внешне пациенты очень напоминают больных с аномалией гена FBN1, но причина их патологии скрывается в других нарушениях. К примеру, гомоцистинурия — это обменное заболевание, которое проявляется системными изменениями соединительной ткани, но может приводить к внезапным инсультам и существенно замедляет умственное развитие ребенка
Поэтому важно точно определить причину заболевания соединительной ткани и своевременно начать лечение.
Прогноз
Современные методы исследования в медицине позволяют выявлять заболевание у детей в раннем возрасте. Это помогает повысить качество жизни таких пациентов и предупредить раннюю смертность. Продолжительность жизни людей с синдромом Марфана при бережном отношении к своему здоровью достигает . Прогноз болезни во многом зависит от выраженности сердечно-сосудистых патологий, поскольку выживание пациентов с этой генетической аномалией определяет состояние аорты и риск ее спонтанного разрыва. Такие люди требуют постоянного наблюдения у врачей различных специальностей для своевременной коррекции проявлений синдрома.










Методы терапии
Прежде всего, лечение заключается:
- в назначении бета-адреноблокаторов;
- в хирургическом вмешательстве при патологиях клапанов и аорты;
- в хирургической коррекции патологий позвоночника.
Из бета-блокаторов пациентам целесообразно назначать препараты в виде пропранолола или атенолола. Это помогает предотвратить серьёзные осложнения со стороны сосудов и сердца. Бета-блокаторы уменьшают интенсивность сокращения сердечной мышцы, снижая её нагрузку, останавливают процесс расслоения аорты и снижают риск развития аневризмы. Если аневризма достигает критических размеров, пациентам показано хирургическое вмешательство.
В качестве консервативного лечения сколиоза, обычно, применяют фиксацию позвоночника, но если он искривлён от 40 градусов и более, операция является более предпочтительным методом.
Всем пациентам, страдающим синдромом Марфана, нужно каждый год проходить обследование у невролога, кардиолога и окулиста, с генетическим консультированием по показаниям.
К каким докторам следует обращаться если у Вас Болезнь (синдром) Марфана:
Кардиолог
Офтальмолог
Кардиохирург
Ортопед
Генетик
Терапевт
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Болезни (синдрома) Марфана, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Что нужно знать медикам о синдроме Марфана?
Поскольку речь идёт о сложном генетическом заболевании, для клиницистов важно чётко формулировать и знать её основные причины, симптомы, принципы диагностики и опасные осложнения. Если обозначить их коротко, они будут выглядеть таким образом:
причина — в генной мутации по аутосомно-доминантному типу, с нарушением синтеза в организме фибриллина-1;
симптомы — разные, клиника широко варьируется, но всегда следует обращать внимание на конечности пациента, состояние его хрусталиков и аорты;
осложнения тоже различны, но самое опасное из них заключается в расслоении аорты;
точно диагностировать синдром Марфана можно только на основании генетического исследования.
Структурные аномалии скелета, глаз и сердечно-сосудистой системы выявляют с помощью диагностической визуализации, а с целью профилактики разрыва аорты следует сразу назначить препараты, относящиеся к группе бета-блокаторов.
Как определить изменения характерные для этой болезни?
Существует ряд признаков, возникающих в результате несовершенства соединительной ткани при синдроме Марфана.
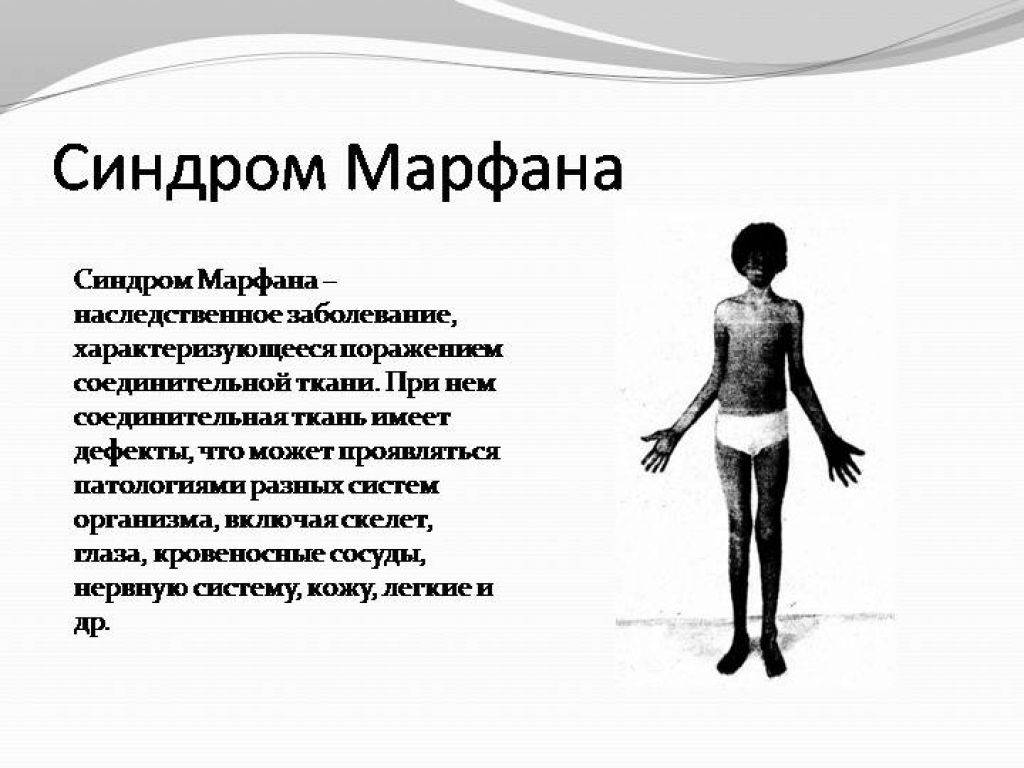
- Высокорослость с периода детства, за счет того, что хрящевая ткань больше поддается процессам вытяжения на фоне слабой соединительной ткани. Интенсивные процессы роста предрасполагают к формированию недостаточно плотной минерализации костной ткани с ранним развитием остеопороза в возрасте после 30 лет.
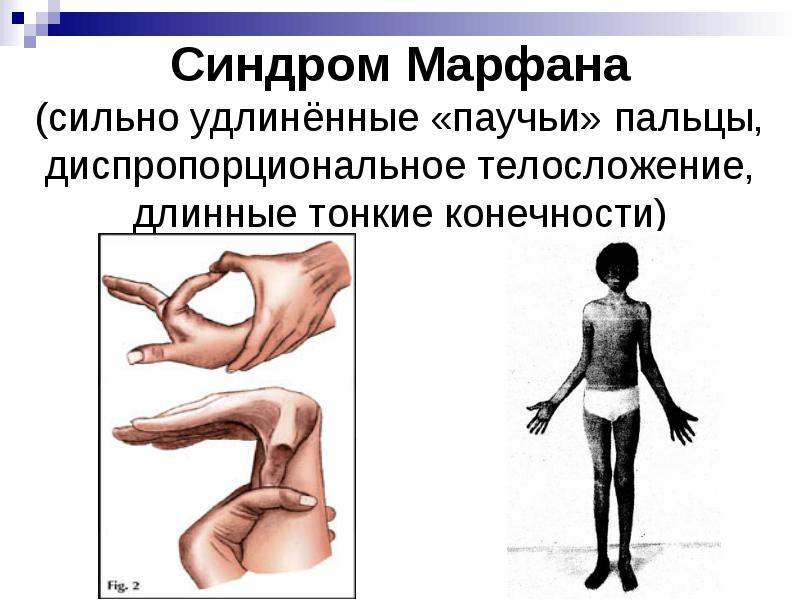
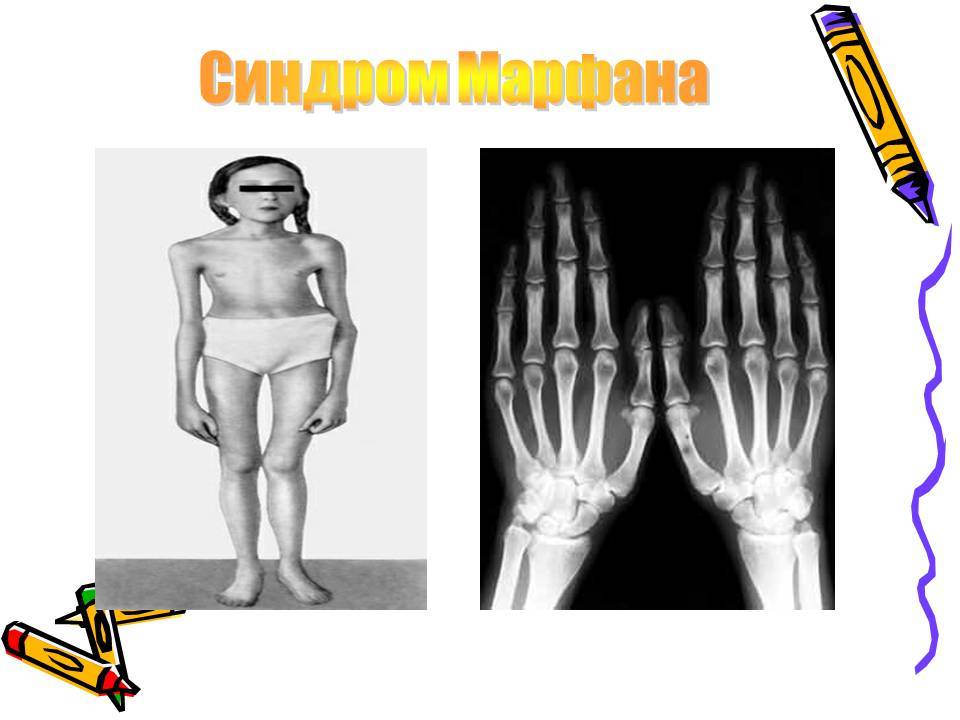
- Астеническое телосложение с непропорционально длинными руками относительно туловища.
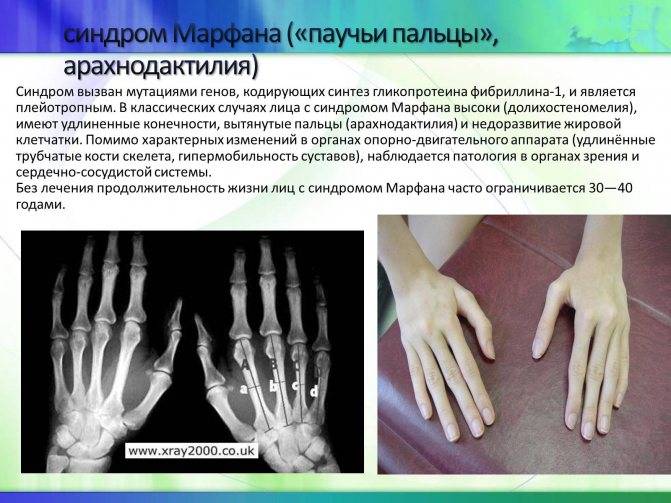
- Типичные пальцы – очень тонкие паукообразные пальцы кистей рук.
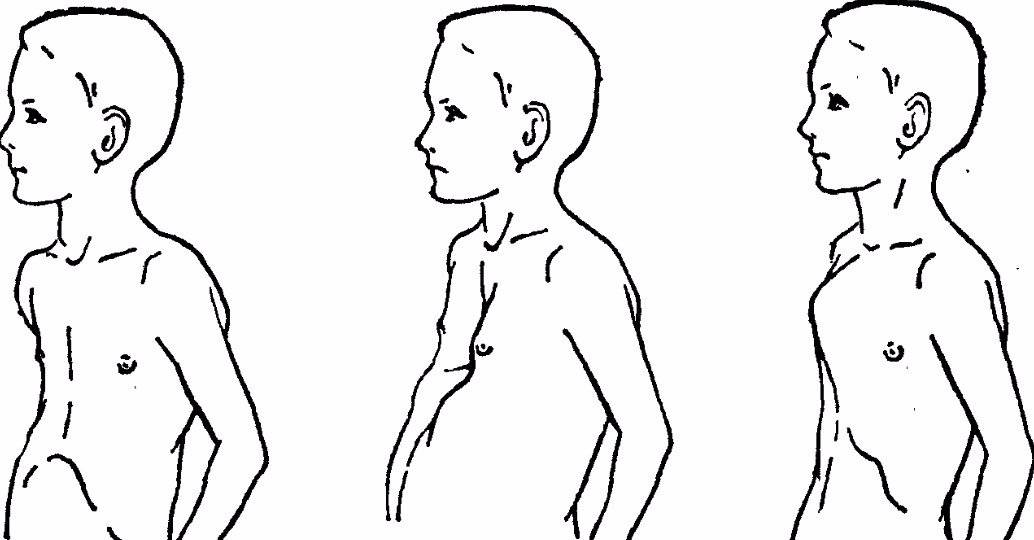
- Деформация грудной клетки – вогнутая грудная клетка.
- Высокое нёбо.
- Нарушение осанки вплоть до формирования кифосколиоза, в результате слабости связочного аппарата позвоночного столба.
- Сверхгибкость – это одно из ярких проявлений синдрома Марфана. Гипермобильность суставов развивается так же за счет слабых связок, расположенных вокруг суставов, что может проявляться их высокой подвижностью в различных отведениях, а так же может провоцировать различные подвывихи.
- Изменения соединительной ткани в области сердца сопровождается пролапсом клапанов сердца, что приводит к нарушению клапанной функции и неполному смыканию клапанов во время сокращения сердечной мышцы. Самое опасное, чем может сопровождаться такая патология – это порок сердца, сопровождающийся недостаточность митрального или аортального клапана. Такие изменения приводят к расширению аорты при неполном замыкании клапана, формируя в дальнейшем аневризму.
- Изменения со стороны зрения проявляются близорукостью высокой степени, которая сочетается с подвывихом хрусталика глаза, развивающегося вследствие слабости цинновой связки.
- Изменения дыхательной системы сопровождаются врожденной патологией легких с недоразвитием легкого, врожденной эмфиземой или бронхоэктазами.
Известные люди с синдромом Марфана
Хоть синдром Марфана – очень редкое заболевание, есть немало знаменитостей, больных синдромом Марфан: Фло Хайман (призер Олимпийских игр по волейболу), Джон Тавенер (композитор), Джоуи Рамон (музыкант), Лесли Хорнби (фотомодель и певица) и другие.
Среди исторических личностей, известных во всем мире, с синдромом Марфана можно выделить:

Музыкант-скрипач Никколо Паганини. Поскольку Паганини умер еще до описания синдрома, данные о его заболевании исследуются по сохранившимся изображениям и дневнику лечащего врача. Скрипач имел характерную деформацию пальцев, высокий рост и худобу, непропорциональное развитие конечностей, впалую грудь, мышечную слабость.



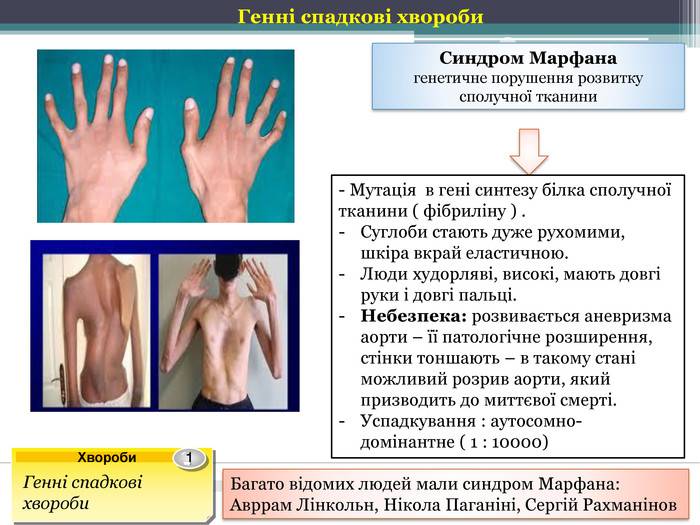


Писатель Ганс Кристиан Андерсен. Имел угловатое лицо, был очень худым и длинноруким, рано заполучил проблемы со зрением.
Президент Америки Авраам Линкольн. Кроме всех внешних признаков синдрома Марфана у Линкольна наблюдались ревматические боли, «разболтанность» суставов, но, в то же время — хорошая физическая выносливость.
Писатель Корней Чуковский. Наличие большого непропорционального носа, длинных конечностей не помешало Чуковскому стать одним из лучших творцов современности и доктором филологии.
Осама Бен Ладен. «Террорист №1» мира имел высокий рост и малый вес и большие проблемы с суставами и позвоночником, а также вытянутый череп и слишком узкое лицо.
Методы диагностики синдрома Марфана
Синдром Марфана диагностируют на основании клинических проявлений, генетического исследования, а также данных эхокардиографии или магнитно-резонансной томографии. С помощью эхокардиографии и МРТ выявляют патологические изменения в области аортального корня и клапанов. Аномальные изменения в хрусталике видны при использовании щелевой лампы, а расширение дурального мешка спинного мозга показывает МРТ. При осмотре глаз окулист видит типичный признак генетической аномалии в виде подвывиха хрусталика.
Тем не менее, в ряде случаев постановка корректного диагноза вызывает трудности, так как многие пациенты не обладают яркими признаками синдрома Марфана. Также у них могут отсутствовать изменения со стороны биохимии и гистологии. В данном случае клиницистам следует ориентироваться на сбор анамнеза, объективный осмотр и генетическое исследование.
Эктопия хрусталика
Эктопия хрусталика глаза — его смещение со своего обычного положения. Из-за растяжения и слабости цинновых связок, на которых он держится, происходит их разрыв. Если повреждение частичное, то хрусталик смещается, но все-таки держится на оставшихся связках — это называется подвывихом. Если же связки Цинна отрываются полностью, то он опускается в полость глаза, свободно меняя свое положение — такое состояние называется вывих.
Основным симптомом этого зрительного нарушения является дрожание радужки — иридодонез. Глубина передней камеры глаза в стороне смещения хрусталика становится более мелкой. Обнаружить иридодонез может врач путем инструментального исследования. Больной со своей стороны испытывает диплопию — двоение в глазах и ухудшение четкости видения. Если вывих произошел в переднюю камеру зрительного органа, то его можно заметить извне — он похож на золотистую каплю масла. При этом в области глаз может отмечаться боль и гиперемия.
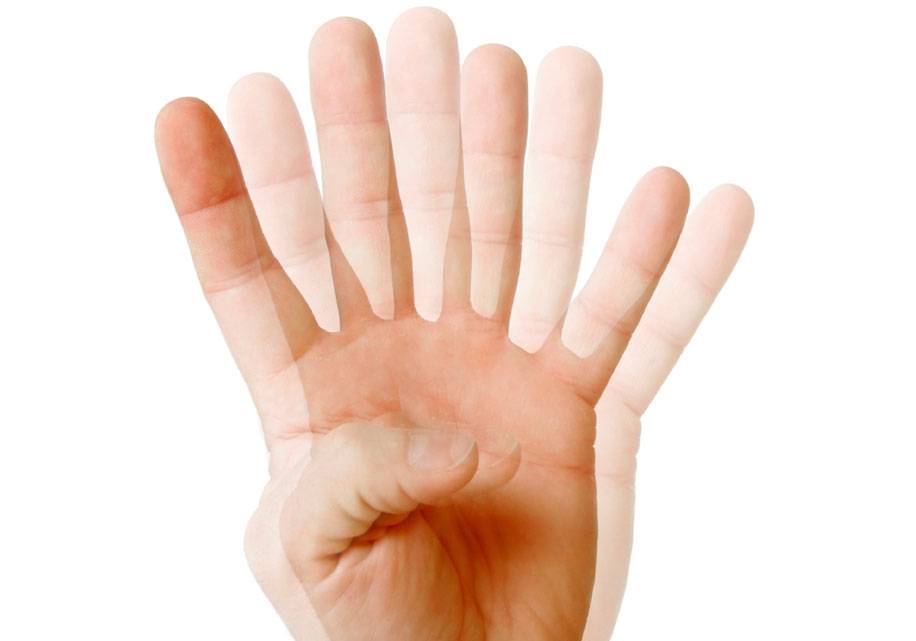
Частичный неосложненный подвывих хрусталика не требует специальной терапии. Для поддержания качества зрения назначаются подходящие контактные линзы. Иногда для его устранения используют способ малоинвазивной транссклеральной фиксации ИОЛ. В поврежденный глаз имплантируют специальное устройство, выполняющее функцию цинновых связок — оно прочно удерживает хрусталик на его анатомическом месте.
1.Общие сведения
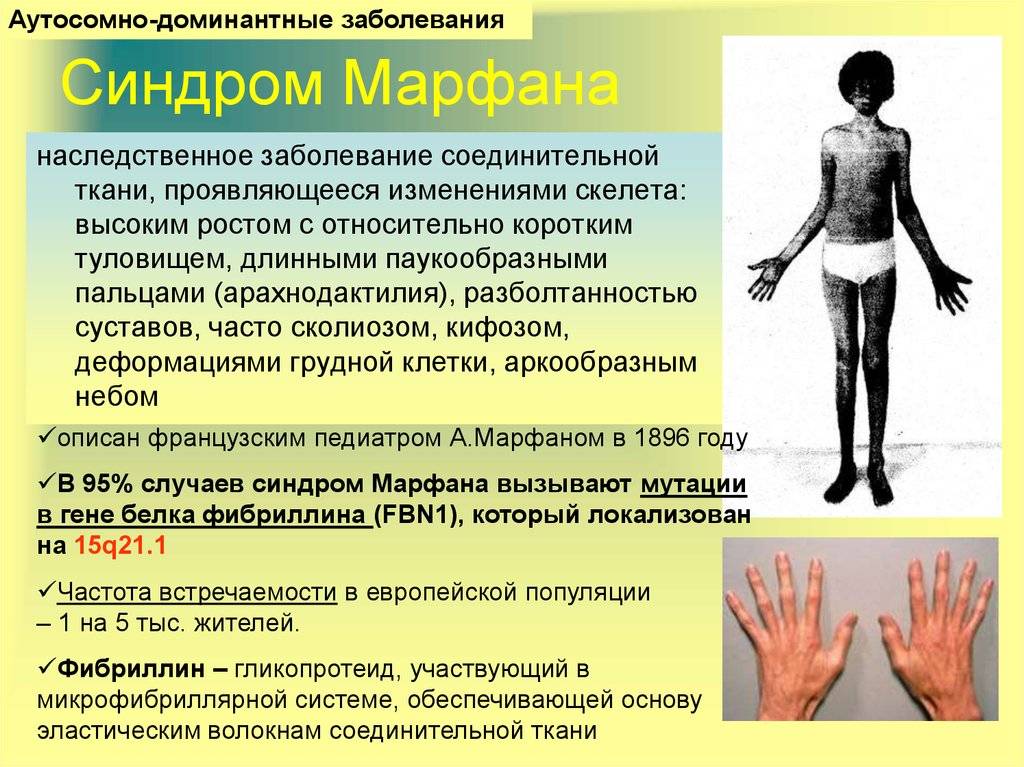
Обычно термины «болезнь» и «синдром», даже если именуются одинаково, в медицине четко разделяются: болезнь представляет собой самостоятельно развивающийся патологический процесс, тогда как синдром (определенная целостная совокупность симптомов) может сформироваться при разных болезнях и по разным причинам. Однако в данном случае «болезнь Марфана» и «синдром Марфана» – это одно и то же: тяжелая генетическая аномалия, врожденное заболевание соединительной ткани, отличающееся ярко выраженной клинической и морфологической спецификой.
Заболевание интенсивно изучается с конца XIX века как классическая наследственная коллагенопатия. Встречается на всех континентах, у лиц любой расы. Среди больных незначительно преобладают лица мужского пола. Относится к редким болезням, хотя эпидемиологические оценки в разных источниках варьируют весьма широко: от 1:5000 до 1:20000.
Возможно, статистической неопределенности способствует то, что отдельные черты (например, арахнодактилия, гигантизм и т.д.) нередко диагностируются как синдром Марфана, в действительности им не будучи.
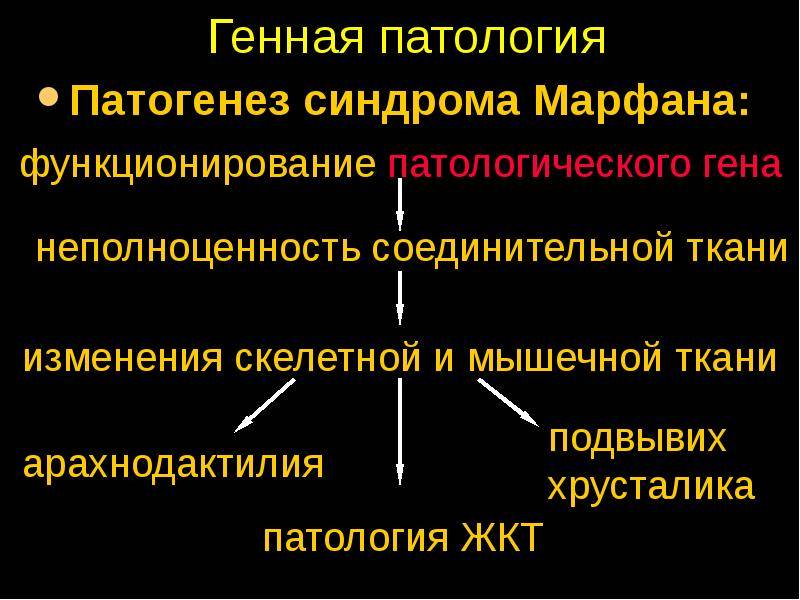
Этиопатогенез
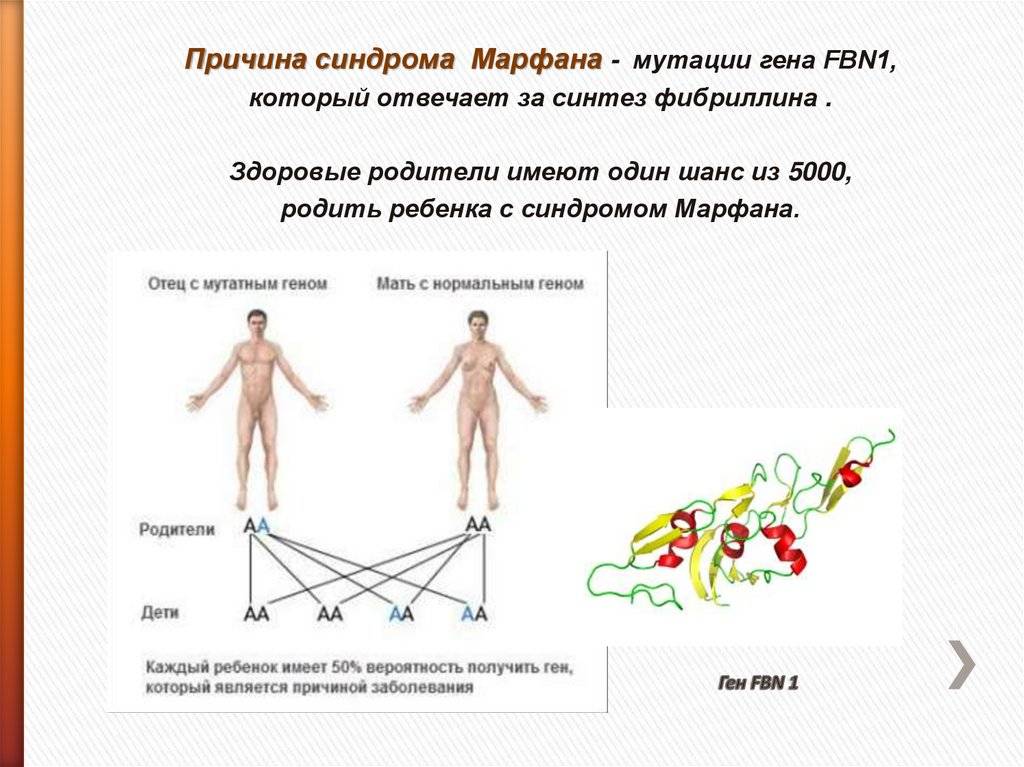
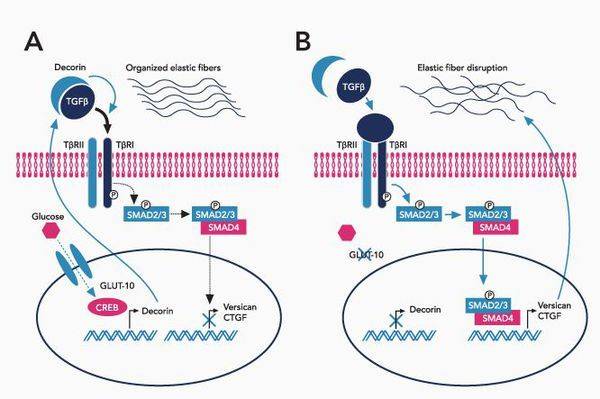
Заболевание отнесено к группе болезней с аутосомно-доминантным типом наследования. Патоморфологическим субстратом синдрома Марфана является нарушение выработки белка фибриллина, который включен в структуру соединительной ткани, обеспечивает ее эластичность и способность к сокращению. Нарушение выработки фибриллина обусловлено мутацией гена FBN1.
Из-за недостатка полноценного фибриллина соединительная ткань становится более растяжимой и не в состоянии выдерживать физиологическую физическую нагрузку.
Синдром Марфана отличается выраженной генетической гетерогенностью.
Причины возникновения

Болезнь Марфана у детей наследуется по аутосомно-доминантному типу (т. е передается от родителя к ребенку).




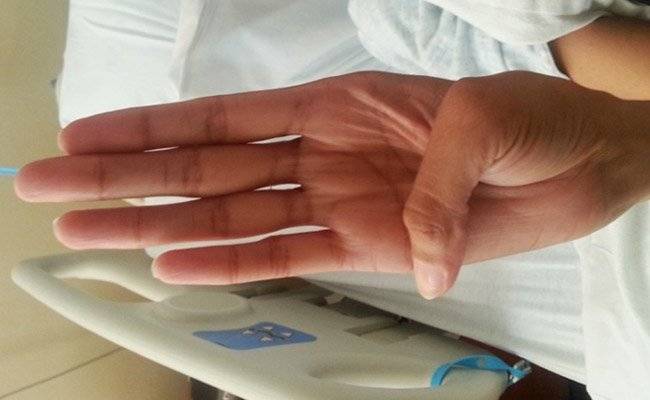

Также возможны мутации за счет воздействия на организм женщины факторов внешней среды (ионизирующее излучение, лучевая терапия, радиация).
Причины возникновения и механизм развития заболевания недостаточно изучены.
Особая роль отводится нарушению процессов обмена, в результате которых накапливается в коллагеновых и эластических волокнах большое количество мукополисахаридов.
Это ведет к тому, что соединительная ткань перерастягивается, легко подвергается механическому воздействию и приводит к развитию клинической симптоматики.
Классификация
Выделяют следующие формы болезни Марфана:
В зависимости от генной предрасположенности:
- семейная (патология передается от родителя к ребенку);
- спорадическая (патология вызвана внезапным мутированием в геноме).
В зависимости от проявлений клиники:
- стертая, когда признаки заболевания практически не проявляются и могут быть не замечены в течение всей жизни. Патологические изменения выявляются в одной или двух системах.
- выраженная, когда признаки заболевания касаются двух и более органов и систем (сердце, кости и суставы, легкие, кожа, глаза).
Классификация синдрома Марфана
Выделяют несколько форм заболевания в зависимости от особенностей клинических проявлений генетической мутации.
Существуют две основные клинические формы патологии:
- Стертая. Таким пациентам «везет» больше: аномалия у них проявляется поражениями только одной-двух систем организма, а симптомы выражены незначительно. Люди могут жить практически нормальной жизнью, несмотря на болезнь.
- Выраженная. В таких случаях поражаются три и более систем организма, либо значительно нарушается функционирование одной из систем.
В зависимости от степени проявления выделяют легкие, среднетяжелые и тяжелые формы синдрома Марфана. Тяжелые патологии встречаются гораздо реже: частота их выявления составляет примерно 1 на 25–50 тысяч человек.
Принципиальную роль в определении прогноза болезни играет характер ее течения:
- Прогрессирующий. В этом случае постоянно появляются новые симптомы заболевания, степень тяжести увеличивается, а с каждым годом жизни пациента возрастают риски фатальных осложнений.
- Стабильный. Такой характер считается наиболее благоприятным: у пациентов со стабильными проявлениями синдрома Марфана клиническая картина практически не меняется на протяжении жизни.
Выделяют три разных, но похожих заболевания:
- Синдром Марфана — стертая форма патологии с положительным результатом генетического тестирования.
- Болезнь Марфана — классическая клиническая картина с подтвержденным семейным наследованием.
- Марфаноподобный синдром — проявление патологии соединительной ткани без генетической мутации.
Первые признаки заболевания чаще всего проявляются еще в детском возрасте. К подростковому периоду становится понятно, насколько быстро у пациента прогрессирует болезнь, вызванная мутацией гена FBN1.
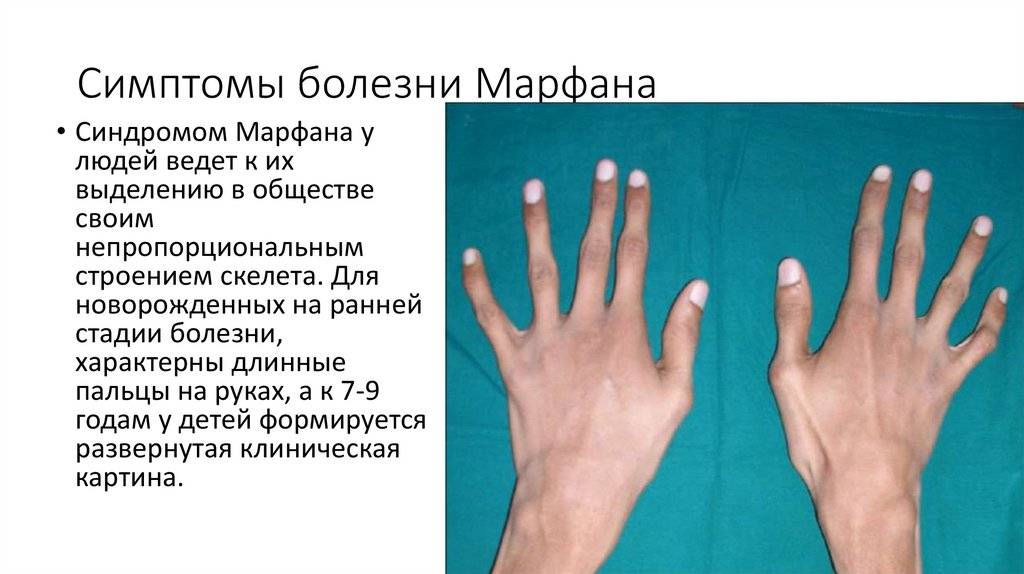
Симптомы болезни Марфана
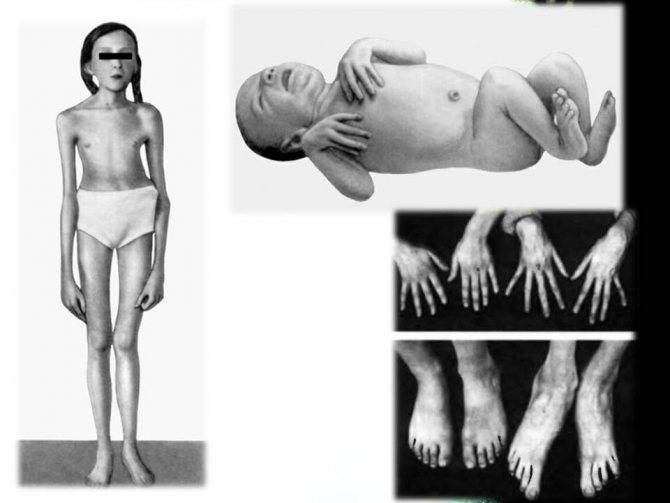
Синдромом Марфана у людей ведет к их выделению в обществе своим непропорциональным строением скелета. Для новорожденных на ранней стадии болезни, характерны длинные пальцы на руках, а к 7-9 годам у детей формируется развернутая клиническая картина.
У взрослых характерна различная симптоматика в зависимости от системы поражения:
- Нервная система: болезненность в поясничной области, головные боли,поражение симпатической и парасимпатической иннервации органов брюшной полости и малого таза (слабость кишечной стенки, недержание мочевого пузыря у ребенка). Также высок риск развития инсульта, субарахноидального кровоизлияния и разрыва аневризм головного мозга.
- Сердечно – сосудистая система: \пороки сердца (сужение легочной артерии, пролабирование створок двустворчатого клапана, дилатационная кардиомиопатия, расширение границ сердца (аорты и всех ее отделов), дефекты МЖП и МЖЖ перегородок. У больных может развиваться нарушение ритма и проводимости в виде аритмий.
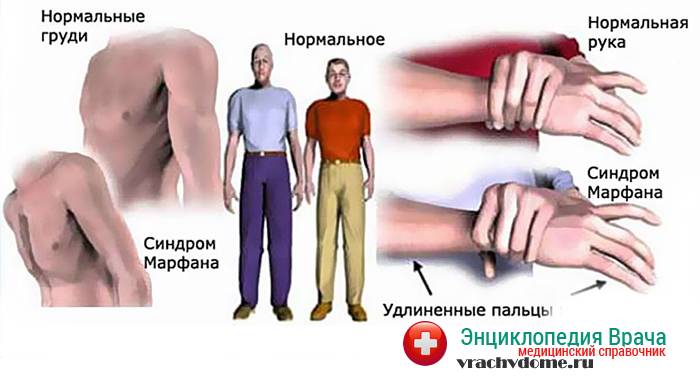
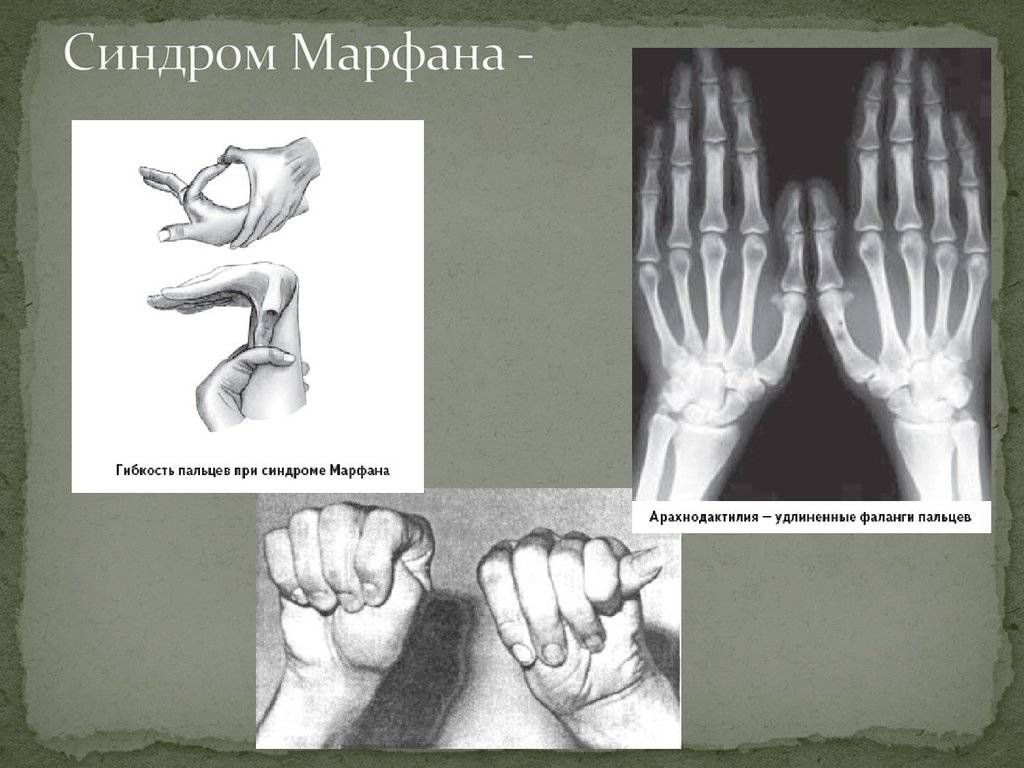
- Опорно-двигательный аппарат: телосложение астенической формы (дети худые), высокий рост у мужчин 190±10 см, у женщин 179±8 см, слаборазвитый подкожно-жировой слой, длинные пальцы (паукообразные), плоскостопие, череп и лицо вытянутые и узкие, недоразвитость скул, нарушение развития зубов и прикуса, вытянутая нижняя челюсть, готическое верхнее небо, гипермобильность суставов (смотрите на картинки выше). С возрастом ребенка может прогрессировать деформация позвоночного столба, с развитием сколиоза. Также может деформироваться грудная клетка, образуется вдавление – «грудь сапожника». Деформированный тазобедренный сустав нередко приводит к инвалидности при неоказанном своевременном лечении.
- Орган зрения: смещение хрусталика за счет слабого связочного аппарата) на ранней стадии, уплощение роговицы, развитие близорукости или дальнозоркости, спазм аккомодации, отслоение сетчатки.
- Кожа и мягкие ткани: перерастяжение кожи с образованием стрий атрофического характера. Они возникают внезапно, не связаны с колебание веса людей, беременностью и гормональным фоном. Кожа липкая, потная, с мраморным оттенком. Подкожно-жировой слой слабо развит, поэтому у больных наблюдаются грыжевые выпячивания в области передней брюшной стенки.
- Дыхательная система: развитие буллезной эмфиземы легких, проявляющейся кашлем, одышкой, развитием дыхательной недостаточности и спонтанного пневмоторакса.
Советы родителям ребенка с синдромом Марфана
Конечно, грустно и горько осознавать, что у ребенка имеется такое опасное заболевание. Однако только правильное поведение родителей поможет преодолеть трудности, в том числе создать нужный психологический настрой. Нужно научиться жить с этой проблемой и постоянно держать под контролем состояние ребенка





Важно найти хороших специалистов, которые будут заниматься его здоровьем
Следует также научить его относиться с достоинством к своему состоянию, не реагировать на возможные насмешки со стороны. Лучше привить ему любовь к занятиям, которыми он сможет заниматься в дальнейшем: пусть это будет программирование или, например, музыка. Если диагноз был поставлен уже в старшем возрасте, то придется отказаться от некоторых видов спорта.

Важно также наладить хороший контакт с учителями, объяснив им, что это заболевание предполагает некоторые особенности. Например, сидеть ему лучше за первой партой из-за проблем со зрением, не стоит испытывать слишком сильных физических нагрузок
Каждому родителю хочется обеспечить своему ребенку счастливое детство. Дети с синдромом Марфана должны знать, что в мире много интересных занятий, которые им доступны.
3.Симптомы и диагностика
Синдром Марфана обнаруживается уже в раннем возрасте; он может прогрессировать или оставаться относительно стабильным. Классической триадой считают деформацию опорно-двигательного аппарата, офтальмологическую патологию и сердечнососудистую недостаточность.
Больные отличаются высоким ростом и диспластичным телосложением: непропорционально длинные конечности, «паучьи пальцы» (арахнодактилия), килевидная или вдавленная грудная клетка, сложные искривления позвоночника (лордоз, кифосколиоз), вальгусная или варусная конфигурация ног, патологическая подвижность суставов, плоскостопие и т.д.
Характерные диспропорции обнаруживаются также в строении челюстно-лицевых структур. Явно недостаточно развиты мышечная и жировая ткани. Типичным признаком является аневризма аорты и/или других крупных сосудов, в т.ч. легочных и мозговых; встречается множество различных пороков сердца. Столь же полиморфными и тяжелыми могут быть аномалии развития органов дыхания, пищеварения, а также центральной нервной и зрительной систем (в частности, офтальмологически выявляется выраженная миопия или гиперметропия, подвывих хрусталика, полное отсутствие радужки или хрусталика, удлинение глазного яблока и т.п.).
Комплекс столь критичных по сути аномалий может быть выражен относительно мягко, однако в целом синдром Марфана обычно нуждается в квалифицированном терапевтическом наблюдении и сопровождении, – в противном случае и качество, и продолжительность жизни резко ограничиваются (в отсутствие курации непосредственной причиной смерти становится, как правило, хроническая или острая, наступающая вследствие разрыва аневризмы, сердечная недостаточность).
Диагноз устанавливается путем изучения семейного анамнеза и всестороннего обследования: практически любой из существующих сегодня диагностических методов выявляет те или иные характерные изменения.
О нашей клинике м. Чистые пруды Страница Мединтерком!
Что провоцирует / Причины Болезни (синдрома) Марфана:
Синдром Марфана – это врожденная аномалия, наследуемая по аутосомно-доминантному типу. Причиной возникновения синдрома являются мутации гена FBN1, который отвечает за синтез фибриллина – структурного белка межклеточного матрикса, предоставляющего эластичность и сократимость соединительной ткани. Аномалия и нехватка фибриллина провоцирует нарушения формирования волокнистых структур и потерю прочности и упругости соединительной ткани, невозможность выдержать физиологические нагрузки. В стенках сосудов эластического типа происходят гистологические изменения. В 75 % случаев синдром Марфана имеет семейный тип наследования, а в 25 % происходит первичная мутация. Если отцу более 35 лет и в его анамнезе есть синдром Марфана, то очень высокая вероятность того, что он передаст заболевание ребенку.