Что такое Синдром Ретта –
Прогрессирующее дегенеративное заболевание ЦНС предположительно генетического происхождения, встречается преимущественно у девочек, названо по имени австрийского ученого A. Rett, впервые описавшего его в 1966 г. Автор сообщил о 31 девочке с регрессией психического развития, аутистичным поведением, утратой целенаправленных движений и появлением особых стереотипных двигательных актов, «сжимания рук».
Распространенность
Частота его относительно высока – 1:10 000 девочек. В мире описано более 20 тыс. случаев заболевания; большинство из них спорадические, менее 1% – семейные. При изучении близнецов показана конкордантность по синдрому Ретта монозиготных и дискордантность дизиготных пар. Географическое распространение синдрома Ретта неравномерно. Отмечено скопление больных в определенных небольших сельских районах «Ретт-ареалы», что может быть связано с существующими популяционными изолятами. Такая концентрация заболевания наблюдается в Норвегии, Италии, Албании и Венгрии.
Признаки и симптомы
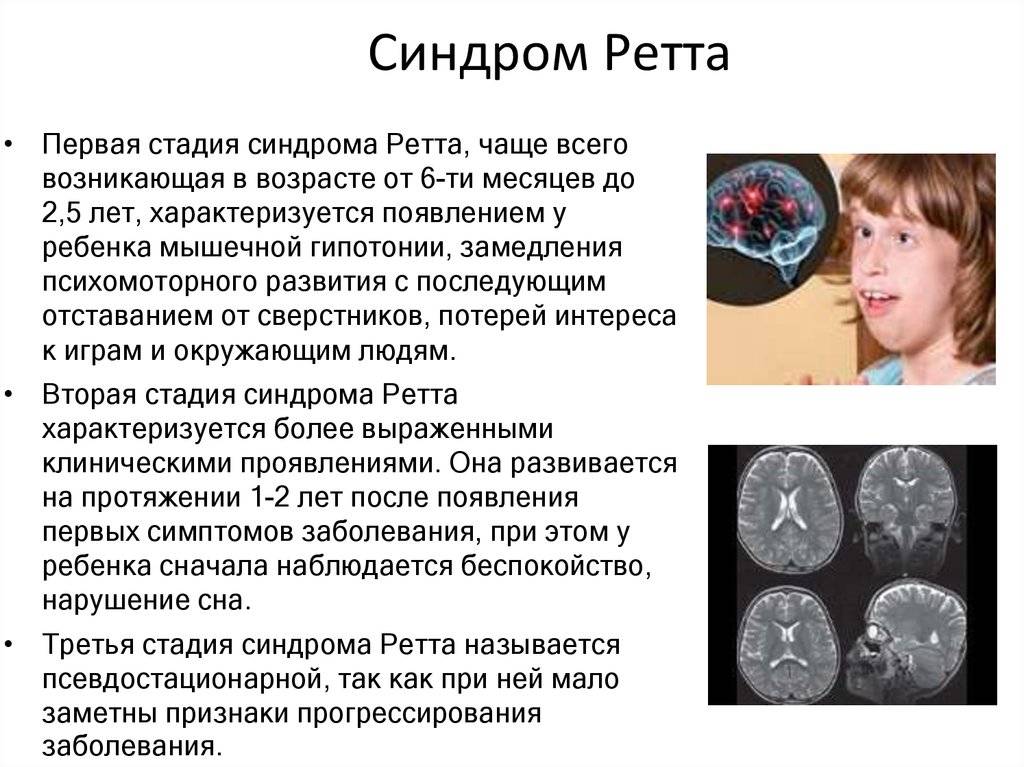
I этап
Стадия I, называемая ранним началом, обычно начинается в возрасте от 6 до 18 месяцев. Эту стадию часто упускают из виду, потому что симптомы расстройства могут быть несколько расплывчатыми, а родители и врачи могут сначала не заметить легкое замедление развития. Младенец может начать меньше смотреть в глаза и потерять интерес к игрушкам. Могут наблюдаться задержки в развитии крупной моторики, такой как сидение или ползание
Может произойти заламывание рук и замедление роста головы, но этого недостаточно, чтобы привлечь внимание. Этот этап обычно длится несколько месяцев, но может продолжаться и более года.
II этап
Стадия II, или стадия быстрой деструкции, обычно начинается в возрасте от 1 до 4 лет и может длиться несколько недель или месяцев. Его начало может быть быстрым или постепенным, поскольку ребенок теряет целенаправленные навыки рук и разговорный язык. Характерные движения рук, такие как заламывание, мытье, хлопанье или постукивание, а также многократное движение рук ко рту часто начинаются на этой стадии, которая называется «ртом». Ребенок может держать руки сцепленными за спиной или по бокам, произвольно касаясь, хватая и отпуская. Движения продолжаются, пока ребенок бодрствует, но исчезают во время сна. Могут возникать нарушения дыхания, такие как эпизоды апноэ и гипервентиляции, хотя дыхание обычно улучшается во время сна. У некоторых девочек также проявляются схожие с аутизмом симптомы, такие как потеря социального взаимодействия и общения. Ходьба может быть неустойчивой, и возникновение двигательных движений может быть затруднено. На этой стадии обычно наблюдается замедленный рост головы.
III стадия
Стадия III, или плато, или псевдостационарная стадия, обычно начинается в возрасте от 2 до 10 лет и может длиться годами. На этой стадии проявляются апраксия , двигательные проблемы и судороги . Однако может наблюдаться улучшение поведения с уменьшением раздражительности, плача и признаков аутизма . На стадии III может появиться больше интереса к окружающему, и могут улучшиться бдительность, концентрация внимания и коммуникативные навыки. Многие девушки остаются на этой стадии большую часть своей жизни.
IV этап
Стадия IV, или поздняя стадия ухудшения моторики, может длиться годами или десятилетиями. Выдающиеся особенности включают снижение подвижности, искривление позвоночника и мышечную слабость, ригидность, спастичность и повышенный мышечный тонус при неправильной позе руки или ноги. Девочки, которые раньше могли ходить, могут перестать ходить. Когнитивные, коммуникативные или ручные навыки обычно не снижаются на стадии IV. Повторные движения рук могут уменьшиться, а зрение обычно улучшается.
Варианты
Признаки и симптомы типичной формы синдрома Ретта хорошо описаны. Помимо классической формы синдрома Ретта, на протяжении многих лет было описано несколько атипичных форм; основные группы:
- Врожденный вариант (вариант Роландо): при этом тяжелом подтипе синдрома Ретта развитие пациентов и окружность их головы отклоняются от нормы с рождения. Типичный взгляд пациентов с синдромом Ретта обычно отсутствует;
- Вариант Заппелла синдрома Ретта или вариант с сохраненной речью: в этом подтипе синдрома Ретта пациенты приобретают некоторые ручные навыки, а язык частично восстанавливается примерно в возрасте 5 лет (то есть после фазы регресса). Рост, вес и окружность головы часто находятся в пределах нормы, при этом наблюдается хорошая грубая моторная функция. Вариант Заппелла – более легкая форма синдрома Ретта;
- Вариант Ганефельда или вариант ранней эпилепсии. При этой форме синдрома Ретта пациенты страдают эпилепсией до 5-месячного возраста.
Само определение синдрома Ретта с годами уточнялось: поскольку атипичные формы существуют близко к классической (Hagberg & Gillgerg, 1993), была введена терминология «комплекса Ретта».
Методы диагностики, применяемые при подозрении на синдром Маркуса-Гунна
Симптоматика синдрома настолько очевидна, что определить его не составляет труда. Однако врачу необходимо выявить причины патологии, тем более если она является приобретенной. Сначала офтальмолог проверяет остроту зрения пациента. После этого проводится исследование глаза посредством щелевой лампы. Данный метод позволяет оценить состояние конъюнктивы, век, роговицы. Рентгенография назначается при травмах. Если есть подозрение, что синдром является следствием поражения центральной нервной системы, пациента отправляют на магнитно-резонансную томографию. Может потребоваться помощь невролога.







Причины
Под этой болезнью понимают изменение (мутацию) гена MECP2 на Х-хромосоме, которая определяет пол человека, вместе с У-хромосомой. Женскому полу характерно две Х-хромосомы, а представители мужского пола одну Х и одну У хромосомы. Болезнь поражает в основном девочек, потому что у них есть вторая копия гена MECP2, которая функционирует, как положено, и ребенок выживает.
Причины происхождения синдрома Ретта не до конца изучены, невзирая на то, что ученые доказали прямое предрасположение наследственности к данной болезни. Поэтому есть несколько предположений, объясняющих развитие синдрома Ретта, но и они до конца не являются точными и верными:
- мутации на генном уровне, спровоцированные высоким количеством родственных связей в поколении (2,7%, при статистике в допустимом формате 0,51%). Появляются они ещё в самом зародыше, когда у плода формируются свои внутренние органы;
- хромосомные несоответствия. Специалисты придерживаются мнения, что проблема обитает в Х-хромосоме, однако выяснить какой именно участок хромосомы патологический не получается;
- метаболические нарушения и дисфункция митохондрий. В клинических случаях специалисты заметили, что у больных с синдромом Ретта наблюдаются изменения в картине крови. Повышается уровень миоцитов и лимфоцитов, также превышают норму молочная и пировиноградная кислота.
На заболевание влияют все факторы, возможно ученые найдут более детальные причины синдрома Ретта. Но патогенез у ребенка остается неизменным — прекращение развития головного мозга, это влияет на рост, развитие внутренних органов, миопатию и развитие сопутствующих болезней.
К каким докторам следует обращаться если у Вас Синдром Ретта:
Психиатр
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Синдрома Ретта, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору – клиника Eurolab всегда к Вашим услугам! Лучшие врачи осмотрят Вас, изучат внешние признаки и помогут определить болезнь по симптомам, проконсультируют Вас и окажут необходимую помощь и поставят диагноз. Вы также можете вызвать врача на дом. Клиника Eurolab открыта для Вас круглосуточно.
Как обратиться в клинику:
Телефон нашей клиники в Киеве: (+38 044) 206-20-00 (многоканальный). Секретарь клиники подберет Вам удобный день и час визита к врачу. Наши координаты и схема проезда указаны здесь. Посмотрите детальнее о всех услугах клиники на ее персональной странице.
Симптомы Синдрома Ретта:
В анте- и перинатальном периодах, в первом полугодии жизни развитие оценивается как нормальное. Однако во многих случаях наблюдаются врожденная гипотония, незначительное отставание в становлении основных двигательных навыков. Начало заболевания от 4 мес. до 2,5 лет, но наиболее часто оно проявляется в возрасте от 6 мес. до 1,5 года. Описывая психопатологический процесс при синдроме Ретта, одни авторы говорят о «дементировании», другие – о неравномерности психических нарушений.
В течении заболевания выделяют 4 стадии:
I стадия (возраст ребенка 6-12 мес.): слабость мышечного тонуса, замедление роста в длину кистей, стоп, окружности головы.
II стадия (возраст 12-24 мес.): атаксия туловища и походки, машущие и подергивающие движения рук, необычные перебирания пальцами.










III стадия: утрата ранее приобретенных навыков, способности к игре, коммуникациям (в том числе визуальным).
IV стадия: распад речи, возникновение эхолалий (в том числе ретардированных), неправильное употребление местоимений.
Первая стадия – стагнация. Включает замедление психомоторного развития ребенка, замедление роста головы, потерю интереса к играм, диффузную мышечную гипотонию.
Вторая стадия – регресса нервно-психического развития – сопровождается приступами беспокойства, «безутешного крика», нарушениями сна. В течение нескольких недель ребенок утрачивает ранее приобретенные навыки, перестает говорить. Что часто ошибочно интерпретируется как аутизм. Появляются стереотипные движения – «мытье рук», их сжимание, стискивание, сосание и кусание рук, постукивание ими по груди и лицу, атаксия и апраксия. Нарушается равновесие при ходьбе, теряется способность ходить. Больше чем у половины детей отмечается аномальное дыхание в виде апноэ до 1-2 мин, чередующееся с периодами гипервентиляции. Дыхательные нарушения отмечаются в период бодрствования и отсутствуют во время сна. У 50-80% девочек с синдромом Ретта возникают эпилептические припадки различных типов, плохо поддающиеся терапии антиконвульсантами. Чаще всего это генерализованные тонико-клонические припадки, комплексные и простые парциальные судороги, дроп-атаки.
После фазы регресса наступает третья стадия – псевдостационарная, охватывающая длительный период дошкольного и раннего школьного возраста. Состояние детей относительно стабильно. На первый план выступают глубокая умственная отсталость, судорожные припадки, экстрапирамидные расстройства по типу мышечной дистонии, атаксии, гиперкинезов. Приступов беспокойства не отмечается.
В конце первого десятилетия жизни начинается четвертая стадия – прогрессирования двигательных нарушений. Больные становятся обездвиженными, нарастают спастичность, мышечные атрофии, вторичные деформации – сколиоз, появляются вазомоторные расстройства преимущественно в нижних конечностях. Характерно отставание в росте без задержки полового созревания. Имеется тенденция к развитию кахексии. Судорожные приступы редкие. У больных с синдромом Ретта на фоне тотального распада всех сфер деятельности наиболее длительно сохраняются эмоциональное общение и привязанности, соответствующие уровню их психического развития.
Причина
Расположение гена, ответственного за синдром Ретта
Генетически синдром Ретта (RTT) вызывается мутациями в гене MECP2, расположенном на X-хромосоме (который участвует в подавлении транскрипции и эпигенетической регуляции метилированной ДНК), и может возникать спорадически или в результате мутаций зародышевой линии. Менее чем в 10% случаев RTT мутации в генах CDKL5 или FOXG1 также напоминают его. Синдром Ретта первоначально диагностируется при клиническом наблюдении, но окончательный диагноз ставится при наличии генетического дефекта в гене MECP2. В некоторых очень редких случаях невозможно найти известный мутировавший ген; возможно, из-за изменений в MECP2, которые не идентифицируются применяемыми в настоящее время методами, или из-за мутаций в других генах, которые могут привести к клиническому сходству.
Утверждалось, что синдром Ретта на самом деле является нарушением развития нервной системы, а не нейродегенеративным состоянием. Одним из свидетельств этого является то, что у мышей с индуцированным синдромом Ретта не наблюдается гибели нейронов, и некоторые исследования показали, что их фенотипы можно частично исправить, добавив функциональный ген MECP2, когда они станут взрослыми. Эта информация также помогла привести к дальнейшим исследованиям, направленным на лечение этого расстройства.
Спорадические мутации
По крайней мере, в 95% случаев синдрома Ретта причиной является мутация de novo у ребенка. То есть он не наследуется ни от одного из родителей. Родители обычно генотипически нормальны, без мутации MECP2.
В случае спорадической формы RTT, мутировавший MECP2, как полагают, происходит почти исключительно в результате мутации de novo мужской копии Х-хромосомы. Пока неизвестно, что вызывает мутации сперматозоидов, и такие мутации редки.
Мутации зародышевой линии
Он также может быть унаследован от фенотипически нормальных матерей, у которых есть мутация зародышевой линии в гене, кодирующем метил-CpG-связывающий белок-2 , MeCP2 . В этих случаях наследование происходит по Х-сцепленному доминантному типу и наблюдается почти исключительно у женщин, поскольку большинство мужчин умирают внутриутробно или вскоре после рождения. MECP2 находится около конца длинного плеча Х-хромосомы в Xq28. Атипичная форма RTT, характеризующаяся инфантильными спазмами или ранним началом эпилепсии, также может быть вызвана мутацией гена, кодирующего циклин-зависимую киназу-подобную 5 ( CDKL5 ). Синдром Ретта поражает одну из 12 500 живорождений женского пола к возрасту 12 лет.








Синдром Маркуса-Гунна: причины офтальмопатологии
Проводя диагностические мероприятия, врач в первую очередь устанавливает природу синдрома Маркуса-Гунна. Причины его бывают врожденными и приобретенными. При этом, когда речь идет о врожденном характере патологии, не подразумевается наследственность. Генетически заболевание никак не обусловлено. Развивается оно спорадически, то есть случайно. Происходит это еще в период внутриутробного развития. Синдром возникает, если формируется аномальное соотношение между тройничным нервом и нервом, который регулирует движения глазного яблока. Факторы, провоцирующие аномалию, точно неизвестны.

Иногда пальпебромандибулярная синкинезия становится следствием родовой травмы. Однако данная форма патологии уже не врожденная, а приобретенная. Помимо родовой травмы, ее причинами могут стать:
- естественное старение организма, сопровождающееся ослаблением мышц, которые отвечают за поднятие век;
- диабетическая невропатия — осложнение при сахарном диабете, характеризующееся повреждением нервных волокон;
- рассеянный склероз;
- синдром Горнера — поражение симпатической нервной системы, проявляющееся на глазах;
- инсульт;
- черепно-мозговая травма;
- глазная мигрень;
- воспалительные болезни головного мозга, включая энцефалит;
- хроническая недостаточность кровообращения — медленно прогрессирующая дисфункция головного мозга;
- сильные потрясение психологического характера;
- побочный эффект, осложнение после процедуры введения ботокса.
Причины синдрома вегетативной дистонии
Синдром вегетативной дистонии классифицируют как следствие разнообразных патологий центральной и периферической нервной системы. СВД не является самостоятельным заболеванием и редко приходит в одночасье. Причины синдрома вегетативной дистонии следующие:
- Проблемы дома, в школе, которые приводят к систематическим постоянным стрессам;
- Поражение головного мозга вследствие проблем протекания беременности;
- Гормональная перестройка в подростковом возрасте (переходный возраст);
- Наследственность, выраженная плохой переносимостью труда, высокой метеотропностью и так далее;
- Заболевания эндокринной системы (сахарный диабет и так далее);
- Бронхиальная астма, язва желудка, гипертония и другие соматические патологии;
- Пассивный образ жизни;
- Систематические заболевания нервной системы;
- Кариозные зубы, гайморит, отит и другие постоянные очаги инфицирования;
- Умственная и физическая перегрузка;
- Постоянные заболевания аутоиммунного типа.
Синдром Маркуса-Гунна — описание болезни
Синдром Маркуса-Гунна, названный так по имени ученого, впервые описавшего болезнь в 1883 году, является офтальмологической патологией. Другое ее название — пальпебромандибулярная синкинезия. Развивается болезнь преимущественно с одной стороны лица. Данная форма диагностируется у 69% пациентов. Двусторонний тип синдрома наблюдается у 31% больных.
Зачастую недуг возникает у людей с врожденным блефароптозом — патологическим опущением века, которое приводит к частичному или полному закрытию зрачка.
Глаз практически постоянно прикрыт веком, из-за чего ухудшается видимость. Органы зрения формируются по этой причине неправильно. Особенно при врожденном типе заболевания. Это чревато серьезными нарушениями зрительных функций.
Блефароптоз при синдроме бывают частичным, когда веко закрывает только треть зрачка, неполным, сопровождающимся перекрытием зрачковой области более чем на половину, и полным, когда зрачка не видно совсем. Кроме того, бывает блефароптоз истинный и ложный. Последний диагностируется при большом скоплении жировой клетчатки под кожей века, а также уменьшении упругости самого глаза.
Такова общая картина синдрома Маркуса-Гунна. Причины возникновения его определяют форму и симптоматику болезни. От этих факторов зависит характер лечения.
Прогноз
Мужчины с патогенными мутациями MECP2 обычно умирают в течение первых 2 лет от тяжелой энцефалопатии , если у них нет одной или нескольких дополнительных X-хромосом или соматического мозаицизма .
Плоды мужского пола с этим заболеванием редко доживают до срока. Поскольку ген, вызывающий заболевание, расположен на X-хромосоме, женщина, рожденная с мутацией MECP2 на ее X- хромосоме, имеет другую X-хромосому с якобы нормальной копией того же гена, в то время как мужчина с мутацией на его X-хромосоме не имеет другая Х-хромосома, только Y-хромосома; таким образом, у него нет нормального гена. Без нормального гена, обеспечивающего нормальные белки в дополнение к аномальным белкам, вызванным мутацией MECP2, плод мужского пола с кариотипом XY не может замедлить развитие болезни, отсюда и неспособность многих плодов мужского пола с мутацией MECP2 выжить до срока .







Однако самки с мутацией MECP2 имеют немутантную хромосому, которая обеспечивает им достаточно нормального белка, чтобы выжить дольше. Исследования показывают, что мужчины с синдромом Ретта могут быть результатом синдрома Клайнфельтера , при котором мужчина имеет кариотип XXY. Таким образом, немутантный ген MECP2 необходим для выживания пораженного Ретта эмбриона в большинстве случаев, а эмбрион, мужской или женский, должен иметь другую Х-хромосому.
Однако было несколько случаев доношенных 46 мужчин с кариотипом XY с мутацией MECP2 (ассоциированной с классическим синдромом Ретта у женщин), которые были поражены неонатальной энцефалопатией и умерли в возрасте до 2 лет. Частота возникновения синдрома Ретта у мужчин неизвестна, отчасти из-за низкой выживаемости плодов мужского пола с мутациями MECP2, ассоциированными с синдромом Ретта, а отчасти из-за различий между признаками, вызванными мутациями MECP2, и признаками, вызванными мутациями Ретта.
Самки могут жить до 40 лет и более. Лабораторные исследования синдрома Ретта могут выявить такие аномалии, как:
- Нарушения ЭЭГ с 2-х летнего возраста
- атипичные гликолипиды головного мозга
- повышенный уровень бета- эндорфина и глутамата в спинномозговой жидкости
- восстановление вещества P
- снижение уровня факторов роста нервов в спинномозговой жидкости
Большая часть смертей происходит внезапно, но у большинства нет установленной причины; в некоторых случаях смерть является наиболее вероятным результатом:
- спонтанная дисфункция ствола мозга
- остановка сердца , вероятно, из-за синдрома удлиненного интервала QT , желудочковой тахикардии или других аритмий
- припадки
- перфорация желудка
Лечение
В современной медицине нет патогенетического лечения синдрома Ретта, и все сводится только к симптоматическому лечению для облегчения состояния больного ребенка. Лечение состоит как из правильного питания и режима, так и из принятия медикаментов. Следующие этапы лечения:
- Специальная диета содержит повышенное количество жиров, именно благодаря им можно поддерживать оптимальный вес ребенка, также частое кормление несколько улучшает и стабилизирует состояние.
- Специальная лечебная физкультура поможет легче справляться с двигательными нарушениями.
- При эпилептических припадках и судорогах, применяются антиконвульсантная терапия. Они способствуют подавлению высвобождения глутамата в центральной нервной системе.
- Для коррекции сна используют мелатонин, который обладает снотворным эффектом.
- Витамины – способствуют улучшению кровообращения в головном мозге, и стимулирует его работу.
- Есть специальные школы и заведения, где проводятся специализированные программы для сохранения речи и коррекции двигательных расстройств. Особенным успокоительным эффектом обладает музыкальная терапия.
Ученые не перестают искать средство для лечения данной патологии, и проводятся опыты на мышах с 15% успехом. Возможно, в скором времени врачи изобретут специфическое лечение.
Предлагаем посмотреть видео, которое рассказывает об особенностях синдрома Ретта и методиках реабилитации детей с этим заболеванием.
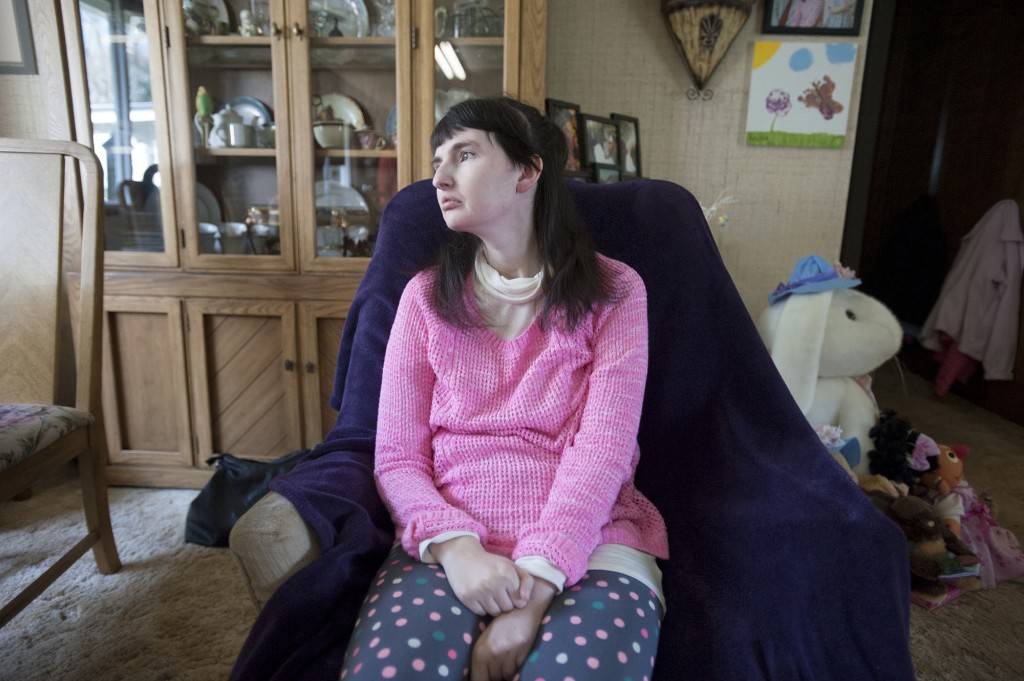
В университет с Tobii Dynavox
 Признаки синдрома Ретта проявились у Джоанны в годовалом возрасте. Мышечный тонус и подвижность стали резко ухудшаться, а в два года болезнь прервала развитие ее речи. Родители видели, что она все понимает, хочет говорить, но не может. Тогда Джоанна сама придумала способ общения, доступный в ее состоянии. Когда девочка с чем-то соглашалась, она энергично мигала глазами, когда хотела сказать «нет» – отводила глаза в сторону. Эта простейшая коммуникация служит ей и сейчас.
Признаки синдрома Ретта проявились у Джоанны в годовалом возрасте. Мышечный тонус и подвижность стали резко ухудшаться, а в два года болезнь прервала развитие ее речи. Родители видели, что она все понимает, хочет говорить, но не может. Тогда Джоанна сама придумала способ общения, доступный в ее состоянии. Когда девочка с чем-то соглашалась, она энергично мигала глазами, когда хотела сказать «нет» – отводила глаза в сторону. Эта простейшая коммуникация служит ей и сейчас.
Сегодня Джо – выпускница католической школы Святой Троицы в Эдмонтоне (канадская провинция Альберта). Она жизнерадостна и независима, несмотря на ограниченные возможности здоровья. Девушка не говорит, но может четко выражать свои мысли. Все это благодаря устройству для коммуникации I-15+ производства Tobii Dynavox, которое управляется взглядом, с языковой программой Sono Scribe. Чтобы управлять ноутбуком и планшетами Джо использует еще одно устройство Tobii Dynavox – PCEye Go.
Учась в начальной школе, Джо использовала разные продукты ААС, включая устройство речевого синтеза, в котором была записана устная речь ее сверстников. В то время она могла поворачивать запястья и с посторонней помощью изолировать указательный палец, поэтому пользовалась словарем для прямого сенсорного прикосновения. Однако болезнь прогрессировала, и целенаправленные движения рук стали невозможными. Тогда Джо и ее родители решили опробовать устройство, которым она бы могла управлять при помощи взгляда. Осенью 2014 года девушка получила PCEye Go производства Tobii Dynavox, а год спустя перешла на устройство I-15+. Джо и ее родители – Симона и Пьер Пикарды – быстро освоили новинки и теперь наслаждаются общением, не прикладывая для этого столько усилий, как раньше.
Кстати, недуг не помешал Джо изучать языки, она, как и все члены ее семьи, свободно общается на английском и на французском. В зависимости от ситуации, Джоанна переключается между английскими и французскими пользователями на своем I-15+.
Джо не просто отлично учиться в школе, увлекается косметологией, общается с друзьями, но и участвует в социальных проектах. Она – член Молодежного совета города Эдмонт и его подкомитета социальной справедливости, а также член-основатель Клуба многообразия у себя в школе. В планах девушки поступление в Канадский университет на политолога. Казалось бы, с синдромом Ретта это невозможно, но коммуникационные устройства Tobii Dynavox сделали мечту реальной.







Лечение
В настоящее время не существует возможности выявлять и влиять на причину этой болезни — мутацию в гене MeCP2. Поэтому синдром Ретта считается неизлечимой болезнью. Хотя ведутся научные работы, посвященные этой теме. Лечению подлежат неврологические осложнения: эпилептические приступы, тремор, спазмы мышц, психомоторное возбуждение. Применяются фармакотерапия (нейролептики, ноотропы), психо-коррекционная работа, логопедическая коррекция, физиолечение, ЛФК и массаж. Важную роль играет правильный уход и надзор за заболевшей.
Клиника РОСА оказывает медицинскую помощь и уход больным с синдромом Ретта. А также психологическую помощь семье и окружению заболевшей.
Симптомы у детей
Симптомы клинически не определяются в первые месяцы жизни ребенка, но в отдельных случаях наблюдается мышечная слабость, начало ползания, сидения и ходьбы немного позже, чем у здоровых детей такого же возраста. Признаки следующие:
- Микроцефалия. Ребёнок рождается с нормальной окружностью головы, но затем она начинает отставать в росте.
- Двигательные нарушения. В период от двух месяцев до пяти лет у ребёнка замечают неспособность удержать бутылочку или игрушки. Также появляются специфические движения — «мытьё рук». Именно этот признак является патогномоничным для синдрома Ретта. Наблюдается хлопанье в ладоши, сжимание, постукивание. Ребёнок кусает руки и стучит ими по телу.
- Умственная отсталость. Все эти изменения можно проследить с помощью психологических тестов. Все те навыки речи и адаптации, которые ребёнок приобрёл, исчезают в течение нескольких месяцев.
- Нарушение координации и их направленность (атаксия и апраксия). Характерен тремор, потеря равновесия, шаткая походка и отрывистые движения.
- Расстройства дыхания. Апноэ (1-2 минуты вплоть до обморока) и гипервентиляции. Во время сна данные симптомы не обнаруживаются.
- Судорожные припадки. Тонико-клинические судороги и эпилептические припадки.
- Сколиоз. Развивается это осложнение из-за гипотонии мышц спины.
Все вышеперечисленные симптомы синдромы Ретта усугубляются с взрослением ребёнка. А если не применять симптоматическое лечение, то состояние будет ухудшаться с невероятной скоростью.